Variant normalization was recently implemented in the hgvs Python package [see blog post]. A major motivation for variant normalization is to facilitate text-based comparisons of variant observations using equivalent canonical representations. From a clinical standpoint, this means we would be better able to compare published variant interpretations.
I apply the normalizer in hgvs package to normalize the variants in Clinvitae (http://clinvitae.invitae.com/) and analyze the results.
The Clinvitae collects and stores clinically observed genetic variants from multiple public databases. All the variants can be downloaded in one file.
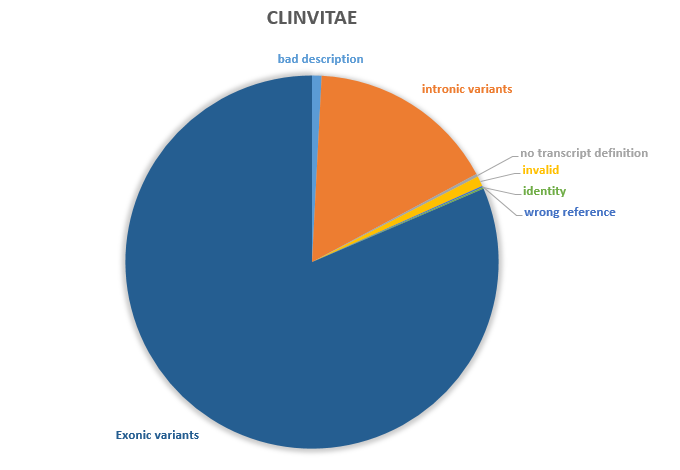
The Clinvitae has 180,974 variant in total, including both genome level and transcripts level variants. In this analysis, I will focus on transcripts level variants (which type is ‘c.’ and the accession starts with ‘NM’). There are a total of 141,338 raw transcripts level variants in Clinvitae. However, 1,162 (0.82%) could not be parsed correctly and are dropped in the following analysis. 23,119 (16.36%) variants are intronic variants and is not supported to be normalized, because the reference sequence of introns is not defined for RefSeq transcripts. 342 variants are defined on sequences unknown to UTA or that do not have genomic alignments in UTA. 1,249 (0.88%) variants are invalid, including variants that base start position greater than end position etc. 183 variants have wrong reference allele. 179 variants are identity variants. For the remaining set of 115,104 (81.44%) variants that could be properly parsed and validated, duplicated variants were removed, reducing the set to 93,761 distinct variants. These distinct variants comprised the test set and was normalized using the hgvs.normalizer module
.
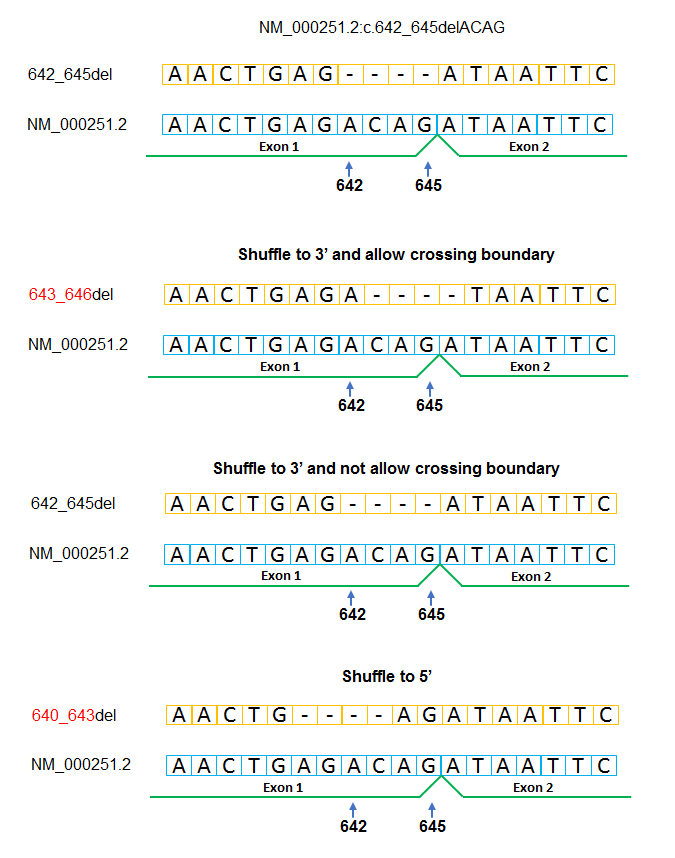
By default, the hgvs normalizer right (3’) shuffles variants and does not permit shuffling across exon boundaries. However, these options are selectable at runtime. To demonstrate this functionality and highly possible errors in variant reporting, we used four configurations during normalization:
- shuffle to 3’ and allow crossing exon-intron boundary
- shuffle to 5’ and allow crossing exon-intron boundary
- shuffle to 3’ and not allow crossing exon-intron boundary
- shuffle to 5’ and not allow crossing exon-intron boundary
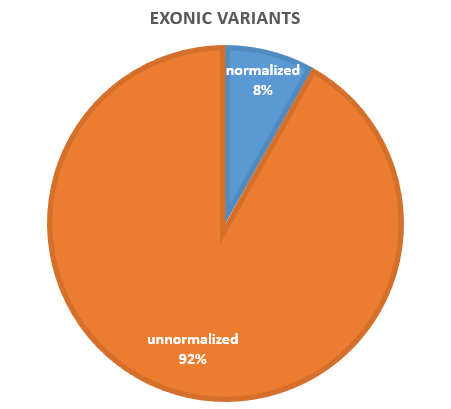
Among these 93,761 variants, 31 variants span the exon-intron boundary. Among these 31 variants, 22 could be normalized when allowing crossing exon-intron boundary. For the remaining 93,730 variants, 86,114 (91.91%) variants remained the same no matter using whichever configuration of normalization, suggesting that these variants are less likely to have other equivalent forms reported 7,586 (8.09%) variants are normalized by at least one configuration.
For the variants that could be normalized, 1,011 could be normalized by 3’ shuffling and 7,020 could be normalized by 5’ shuffling. This indicates that most variants in Clinvitae have already located at 3’ most, consistent with the HGVS recommendations. 445 variants could be shuffled to either 3’ or 5’ direction.
27 variants are located at 3’ end of exons, so they could be 3’ shuffled only when allowing crossing exon-intron boundary and remained the same when that is not allowed. Two variants located near the 3’ end of exons normalized to different results depending on whether exon boundary crossing was permitted. Similarly, 62 variants locate at 5’ end of exons could be 5’ shuffled when allowing boundary crossing and remained the same when that is not allowed. 40 variants located near 5’ end of an exon and the normalization results are different for crossing exon-intron boundary is allowed or not. Other variants normalization results are not affected by whether allowing crossing exon-intron boundary.
The hgvs normalizer also rewrites variants according to HGVS recommendations depending on sequence context. In the test set, 169 insertions were converted to duplications and 22 delins were converted to inversions.
In summary, some variants in Clinvitae are not normalized as 3’ most as possible according to the HGVS recommendations. And some variants are not correctly described. By using a standard, freely-available normalization process, we will be able to more reliably correlate variant observations with clinical significance.
Attachments
Raw results of parsing validating and normalizing for transcript variants in Clinvitae
Clinvitae transcript variants that could be normalized by at least one configuration
Raw results of parsing validating and normalizing for transcript variants in Clinvitae
Clinvitae transcript variants that could be normalized by at least one configuration
i like it! it's a good job!
ReplyDelete